


Algunas Consideraciones sobre la Gestión de Riesgos según la ISO 14971 para los Dispositivos Médicos en Perú
Q.F. Walther Ricardo Vicente Mallma
Associate Consultant and Medical Devices Regulatory Affairs Coordinator
SCR Consulting Peru
RESUMEN
El reporte de gestión de riesgos se constituye como uno de los requisitos regulatorios para el registro de dispositivos médicos en Perú. De acuerdo al Decreto Supremo 016-2011-SA: Reglamento para el Registro, Control y Vigilancia Sanitaria de Productos Farmacéuticos, Dispositivos Médicos y Productos Sanitarios y sus posteriores modificaciones, dicho documento debe estar acorde según norma ISO específica vigente, siendo la ISO 14971 lo exigible actualmente. El presente artículo busca brindar alcances de cómo abordar dicho documento, dando a conocer las etapas que están inmersas en el proceso de la gestión de riesgos, haciendo énfasis en la importancia que supone dicho documento en la salud de las personas.
Palabras clave: Riesgo, peligro, situación peligrosa, daño.
Tal como su nombre lo indica, el estándar internacional ISO 14971: Dispositivos Médicos - Aplicación de la Gestión de Riesgos a los Dispositivos Médicos nos brinda una orientación y dirección respecto al proceso de gestión de riesgos en dispositivos médicos, en el presente artículo abordaremos algunos aspectos relevantes que nos permitirá tener un mejor entendimiento de dicho estándar, así como de la relevancia que ello tiene en relación con la salud pública nacional.
Es por ello, imprescindible el tener claro el concepto exacto del término “riesgo”. La Real Academia Española (RAE), lo define como la “contingencia o proximidad de un daño”; por otro lado, la ISO 14971 lo define como la combinación de la probabilidad de ocurrencia de un daño y la gravedad de dicho daño; y finalmente la normativa peruana, de acuerdo al Decreto Supremo 003-2020-SA: Reglamento que establece las reglas de clasificación y los principios esenciales de seguridad y desempeño de los dispositivos médicos, lo define como la combinación de la probabilidad de que ocurra un daño y la severidad de dicho daño. De acuerdo a dichas definiciones, se observa que la definición para la regulación peruana es la misma que la de la ISO 14971, por lo que para fines regulatorios se adoptará dicha definición.
El no poder estimar o controlar un posible riesgo, puede acontecer en eventos perjudiciales en los pacientes, pudiendo ser alguno de ellos algún familiar, conocido o nosotros mismos. Es allí, donde radica la importancia que tiene la gestión de riesgos en la salud pública. La intención de la gestión de riesgos es la de identificar, evaluar, analizar, valorar y mitigar posibles problemas de los dispositivos médicos; cada una de estas etapas las estaremos revisando detalladamente en los siguientes apartados.
Tal como se puede apreciar en la Ilustración 1: Visión General del Proceso de Gestión de Riesgos, el proceso de la gestión de riesgos se compone de 7 etapas, las cuales no necesariamente tienen solo un inicio y un final, sino que estas etapas son cíclicas ya que se pueden encontrar o suscitar nuevos riesgos durante la etapa de producción o la etapa de comercialización del dispositivo médico.
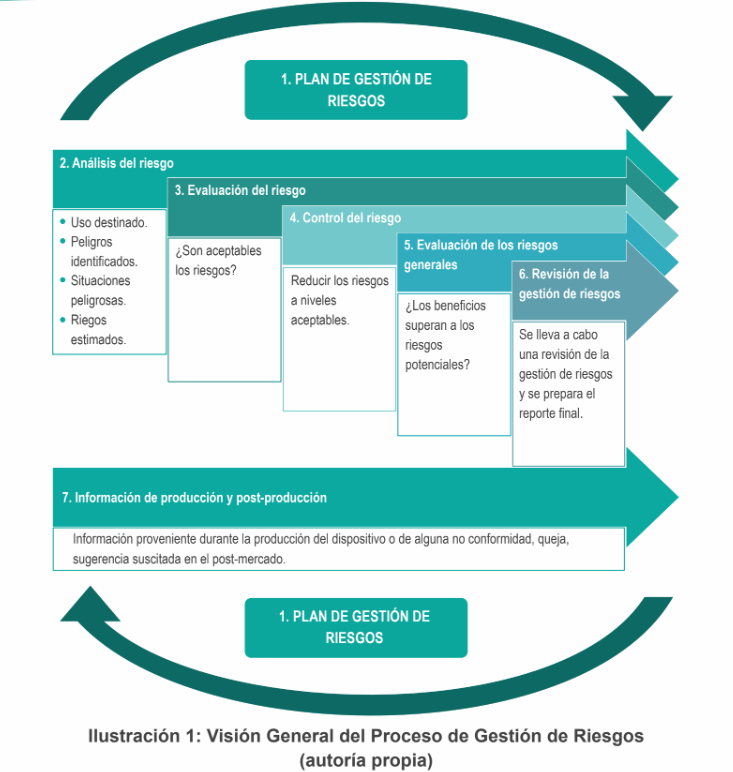
(autoria propia)
- 1. Plan de gestión de riesgos
Identifica principalmente las actividades a realizar en la gestión de riesgos durante el ciclo de vida del producto. Así mismo, define los roles y responsabilidades de cada miembro encargado de elaborar el documento de gestión de riesgos, así como establece los criterios a ser empleados en cada etapa del proceso de gestión de riesgos.
- 2. Análisis de riesgo
Como primer paso para realizar el análisis de riesgo, es importante definir el uso que tendrá el dispositivo médico, ya que sobre dicho uso se identificará los peligros asociados a su uso, incluidos los usos inadecuados.
Para la identificación de peligros es importante conocer las indicaciones para el uso del dispositivo, así se puede identificar potenciales peligros en cada etapa de uso. Estos peligros generalmente se enfocan en los daños al paciente; sin embargo, también se debe considerar los daños a los usuarios, el lugar de uso y el medio ambiente.
Así como la identificación de peligros, la identificación de situaciones peligrosas es crucial en el análisis. Estas situaciones peligrosas son antecedidas por una secuencia previsible de eventos. Por ejemplo, una situación peligrosa podría ser una infección generalizada en un paciente y en este caso particular la secuencia de eventos podría iniciar en una mala manipulación o mal almacenamiento de un dispositivo para cirugía, el cual es estéril, conllevando a una contaminación del dispositivo, lo que finalmente deriva en la infección del paciente. Cada situación peligrosa puede tener múltiples peligros.
Finalmente, a cada peligro que haya sido identificado, se necesita estimar su riesgo. Es así que, para realizar la estimación de los riesgos, se suele definir descripciones para varios niveles tanto para la severidad como para la ocurrencia (ver Tabla 1: Niveles de severidad y ocurrencia); estas descripciones son definidas por el fabricante. Recordemos que los riesgos se definen como la combinación de la probabilidad de que ocurra un daño y la severidad de dicho daño. Table 1: Severity and occurrence levels); these descriptions are defined by the manufacturer. Remember, risks are defined as the combination of the probability of harm occurring and the severity of that harm.

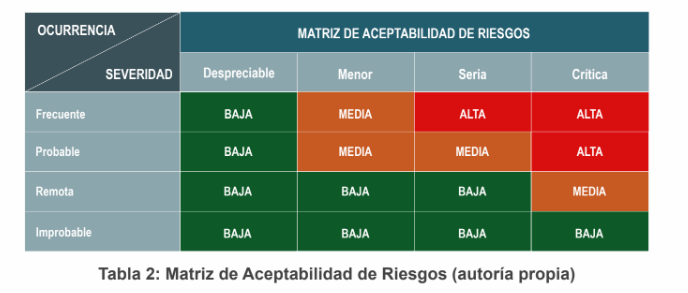
Finalmente, la estimación final se realizará cruzando la información tanto de la severidad como la ocurrencia y ello se puede realizar empleando una Matriz de Aceptabilidad del Riesgo, en donde finalmente se estiman los riesgos como bajos, medios o altos o con otra categoría que asigne cada fabricante. La siguiente tabla (Tabla 2: Matriz de Aceptabilidad de Riesgos), nos muestra un ejemplo de la matriz mencionada empleando los criterios adoptados en la Tabla 1:
- 3. Evaluación de riesgo
En esta etapa toca evaluar si los riesgos son aceptables o si requieren una reducción del riesgo. Generalmente las zonas verdes de la matriz de aceptabilidad de riesgos se suelen definir como aceptables, las zonas rojas como no aceptables y las zonas naranjas como tan bajas como razonablemente pueden ser, conocido generalmente con la terminología ALARP(Tan Bajo como sea Razonablemente Factible). Cabe mencionar que, en la normativa europea, de la que generalmente se basa nuestra normativa, se exige reducir los riesgos tanto como sea posible. Esto es, que inclusive los riesgos evaluados como aceptables tienen que ser considerados en ser reducidos aún más.
- 4. Control de riesgos
Las medidas a realizar para reducir los riesgos no aceptables, se denominan como control de riesgos. El control de riesgos debe tener un impacto más significativo en la probabilidad de ocurrencia del daño ya que la reducción en la severidad de un peligro es más plausible en la práctica. Mucho de las medidas adoptadas en el control de riesgos se ven reflejadas en adiciones de advertencias o precauciones en los manuales de instrucción de los dispositivos médicos.
Luego de implementar los controles de riesgo, se debe evaluar los riesgos residuales para ver si todos ellos son aceptables y en caso alguno no lo sea se debe identificar otros medios para reducir dichos riesgos. No obstante, es posible que haya riesgos que aún permanezcan no aceptables y es en donde se debe realizar un análisis riesgo-beneficio, en donde se debe considerar un riesgo no aceptable si en caso el beneficio supera al riesgo, a través de evidencia objetiva sin razones financieras.
5. Evaluación de los riesgos generales
Etapa en donde se evalúa si el riesgo general del producto es aceptable o no aceptable; ello en base a la evaluación individual de cada uno de los riesgos.
- 6. Revisión de la gestión de riesgos
Etapa en donde se realiza una revisión de todas las actividades del proceso de gestión de riesgos, donde finalmente se plasmará todos los resultados en un Reporte de Gestión de Riesgos.
- 7. Información de producción y post-producción
Tal como se comentó inicialmente, el proceso de gestión de riesgos es uno cíclico, para términos prácticos es un documento vivo que constantemente tiene que ser actualizado en base a nueva información adquirida en los procesos de producción o en el mercado mismo. La identificación de peligros en un inicio se realiza en base al diseño del dispositivo médico, pero de forma general, casi siempre, dichos peligros suponen solo dos tercios de los peligros ya que el otro tercio suele aparecer durante la etapa post-mercado en donde las situaciones ya no son las de los laboratorios.
Tras la revisión realizada al proceso de gestión de riesgos según la ISO 14971 para los dispositivos médicos en el Perú, se evidencia que nuestra normativa se ampara en estándares internacionales, lo cual denota un interés genuino para la salud pública en el país, ya que los dispositivos médicos que ingresen al Perú cumplirán con las condiciones mínimas necesarias que garantizarán la seguridad de los mismos. Como se pudo apreciar en el presente análisis, el conocimiento de la estructura de la ISO 14971 (Dispositivos Médicos - Aplicación de la Gestión de Riesgos a los Dispositivos Médicos) es de vital importancia para cumplir con el requerimiento regulatorio solicitado por la DIGEMID.