


Aspectos Regulatorios para el Reacondicionado de PF y DM en Perú
Q.F. Milagros Consuelo Arista Arévalo
Quality and Regulatory Affairs Specialist
SCR Consulting Peru
Teniendo como objetivo establecer las condiciones técnicas y sanitarias para el funcionamiento de los Establecimientos Farmacéuticos, en julio del 2011 se publicó en el Diario Oficial El Peruano, el “Reglamento de Establecimientos Farmacéuticos” aprobado a través Decreto Supremo N° 014-2011-SA. En este documento se señalan por primera vez, de manera detallada, los aspectos referidos al reacondicionado de productos farmacéuticos y dispositivos médicos.
Es entonces que se define al reacondicionado, de acuerdo al D.S. N° 014-2011-SA y sus posteriores modificatorias, como el conjunto de operaciones al que es sometido un producto terminado nacional o importado, el cual puede consistir en:
- Colocar al mismo producto en un nuevo envase (mediato o inmediato).
- Inclusión o cambio de inserto.
- Agregar información en el envase (primario o secundario) cuya impresión debe ser clara, legible e indeleble a efectos que pueda contar con lo requerido en su registro sanitario.
En tanto que según el Decreto Supremo N° 029-2015-SA: Modificación del Reglamento para el Registro, Control y Vigilancia Sanitaria de Productos Farmacéuticos, Dispositivos Médicos y Productos Sanitarios, para el caso de dispositivos médicos, el reacondicionado hace referencia a agregar información en el envase mediato o inmediato cuya impresión debe ser clara, legible e indeleble, un cambio de inserto o manual de instrucciones a efectos que pueda contar con la información requerida en su registro sanitario.
Antes de continuar este análisis sobre el reacondicionado, es importante hacer una marcada diferencia entre el concepto de reacondicionado propiamente, mencionado líneas arriba, y los conceptos de fraccionamiento y acondicionado.
Considerando nuevamente al “D.S. N° 014-2011-SA y modificatorias”, en este documento se señala que el acondicionado es definido como las operaciones a las que tiene que ser sometido un producto que ya se encuentra en su envase inmediato, para que se convierta en un producto terminado. Mientras que, por otro lado, esta normativa menciona que el fraccionamiento viene a ser la división del contenido de un todo, sea materia prima, producto farmacéutico, dispositivo médico o producto sanitario, que lo contenga, realizado en un laboratorio autorizado para tal fin. Se debe tener claro en este punto que el fraccionamiento no incluye reacondicionado.
Las droguerías pueden encargar a los laboratorios de productos farmacéuticos, dispositivos médicos o productos sanitarios el servicio de reacondicionado, los mismos que deben contar con certificación de Buenas Prácticas de Manufactura (BPM) o documento equivalente emitido por la Autoridad o Entidad competente, según corresponda; de acuerdo al Artículo 71° del Decreto Supremo N° 016-2019-SA: Modificación del Reglamento de Establecimientos Farmacéuticos.
Asimismo, según lo señalado en el anteriormente citado Decreto Supremo N° 016-2019-SA, las droguerías que encarguen el servicio de reacondicionado deben solicitar autorización sanitaria a la Autoridad Nacional de Productos Farmacéuticos, Dispositivos Médicos y Productos Sanitarios (ANM), en el ámbito de Lima Metropolitana, o a la Autoridad Regional de Salud (ARS) correspondiente, a través de la Autoridad de Productos Farmacéuticos, Dispositivos Médicos y Productos Sanitarios de nivel Regional (ARM).
De la misma normativa menciona anteriormente, en cuanto al encargo o ampliación del servicio de reacondicionado a laboratorios nacionales, la droguería debe solicitar autorización sanitaria; para lo cual se debe presentar a la DIGEMID, los siguientes documentos:
- a) Solicitud con carácter de declaración jurada, la cual se descarga desde la página web de la DIGEMID.
- b) Copia del contrato entre las partes relacionado al servicio a brindar, de acuerdo a lo establecido en el Manual de BPM.
- c) Copia del certificado de BPM del laboratorio nacional o extranjero que brinda el servicio de fabricación otorgado por la DIGEMID.
- d) Listado de productos a reacondicionar:
- En el caso de productos farmacéuticos se debe consignar en el listado el nombre del producto, la Denominación Común Internacional (DCI), concentración y forma farmacéutica a reacondicionar.
- En el caso de Dispositivos Médicos se debe consignar en el listado el nombre del producto o dispositivo y su clasificación.
Asimismo, los laboratorios también pueden encargar a otros laboratorios, previa autorización de la DIGEMID la fabricación de productos o dispositivos, sea en su totalidad o en algunas etapas del proceso de manufactura como el reacondicionado; presentando los documentos señalados anteriormente, según lo señalado en el Artículo 109° del Decreto Supremo N° 016-2019-SA.
Según este último Decreto, se precisa que la responsabilidad por la calidad del producto o dispositivo médico, es asumida solidariamente por el titular del registro sanitario y del laboratorio fabricante, por lo que podemos evidenciar la importancia de un trabajo colaborativo y comprometido entre ambas partes.
En junio del 2013, la DIGEMID publicó el Comunicado “Reacondicionado de productos farmacéuticos, dispositivos médicos y productos sanitarios”, siendo que a través de este documento la entidad regulatoria autoriza el servicio de reacondicionado de productos a través Inkjet o de Etiquetas. Con este comunicado, la DIGEMID, también señala que, para el reacondicionado de productos farmacéuticos y dispositivos médicos, se podrá agregar la siguiente información:
- A través de Inkjet: Datos del importador, registro sanitario, nombre del Director Técnico (D.T.), nombre del laboratorio que reacondiciona.
- A través de Etiqueta (Sticker): Datos del importador, nombre del D.T.
Asimismo, en el Comunicado citado anteriormente se señala, los datos o información que no se autorizan como servicios de reacondicionado, los cuales son:
- Nombre del Producto
- Nombre y País del Laboratorio Fabricante
- Número de Lote
- Fecha de Vencimiento
- Denominación Común Internacional
- Concentración.
Sin embargo, para el caso de dispositivos médicos importados cuyo rotulado no consigne, de origen, el nombre del fabricante en concordancia con la Resolución Directoral de inscripción o reinscripción, la DIGEMID señala que permitirá el reacondicionamiento para inclusión del nombre del fabricante (legal o real, según corresponda).
Por otra parte, para el año 2017, de acuerdo al Decreto Supremo N° 016-2017-SA: Modificación del Reglamento para el Registro, Control y Vigilancia Sanitaria de Productos Farmacéuticos, Dispositivos Médicos y Productos Sanitarios, se dispone que ya no se requerirá consignar el nombre del laboratorio que realiza el reacondicionado, así como se podrá exceptuar la inclusión del nombre del Director Técnico en los rotulados de los envases mediatos e inmediatos de los productos farmacéuticos y dispositivos médicos.
En ese sentido para el reacondicionado de productos farmacéuticos y dispositivos médicos, se podrá agregar la siguiente información:
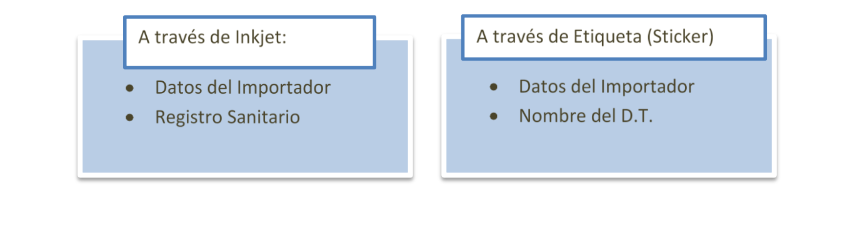
En la práctica como parte del sistema de aseguramiento de la calidad, los laboratorios nacionales que realizan el reacondicionado de los productos farmacéuticos o dispositivos médicos solicitan, normalmente, a las droguerías o laboratorios solicitantes del servicio, los siguientes documentos:
- La orden del servicio de reacondicionado.
- Listado de productos a reacondicionar.
- Registro Sanitario de los productos a ser reacondicionados.
- Certificado de Análisis o Certificado de Conformidad de los productos a reacondicionar.
Mientras tanto las droguerías o laboratorios solicitantes piden a sus proveedores de servicio de reacondicionado, los Reportes de inspección del producto terminado.
Actualmente para el encargo o ampliación del servicio de reacondicionado, los laboratorios y droguerías realizan una Comunicación de Encargo de Servicio de Reacondicionado a la DIGEMID, para lo cual presentan los mismos requisitos detallados en el D.S. N° 016-2019-SA, utilizando el Formato de Orientación como solicitud con carácter de declaración jurada el cual se descarga desde la página web de la DIGEMID y exceptuando el comprobante de pago, puesto que este trámite no tiene costo. En ese sentido, al ser una comunicación, la DIGEMID no emite una Resolución Directoral de Autorización. Como sustento de la autorización y como evidencia en auditorías, las droguerías o laboratorios archivan la documentación presentada a la Autoridad e imprimen la “Consulta de Expediente” en situación de ATENDIDO del Sistema de Trámite Documentario de la DIGEMID.
Cabe resaltar que las droguerías o laboratorios pueden tener uno o más proveedores que les brinde el servicio de reacondicionado de productos, para lo cual deberán realizar la respectiva Comunicación a la DIGEMID para cada proveedor por separado cumpliendo con los requisitos antes mencionados para cada uno.
Es importante recordar que las droguerías y laboratorios que infringen en fabricar, importar, almacenar, distribuir, comercializar o dispensar productos o dispositivos médicos, sin consignar o modificar en el rotulado del envase mediato o inmediato información técnica aprobada en el registro sanitario u otra información exigida en las normas sanitarias de rotulado, o consignando información técnica no autorizada, con excepción del número de lote, fecha de expiración, y de aquellos productos o dispositivos médicos a los que se les autorizó información de acuerdo al registro sanitario o en el agotamiento de stock, tendrán una sanción que asciende a los 3UIT, esto de acuerdo a lo establecido en el Decreto Supremo N° 029-2015- SA.
De la evaluación mencionada se puede observar que, en los últimos años, en Perú, se han venido detallando cada vez mejor los lineamientos y procesos para el reacondicionado de productos farmacéuticos y dispositivos médicos, a través de las normas técnicas nacionales, lo cual permite a los responsables del aseguramiento de la calidad de las droguerías y laboratorios implantar un sistema de gestión de calidad más robusto y garantizar la calidad de los productos entregados a la población.